Somatic Mosaicism and Rare Diseases
Somatic Mosaicism and Rare Diseases
by Hoi Kiu Wong
Introduction to Mosaicism
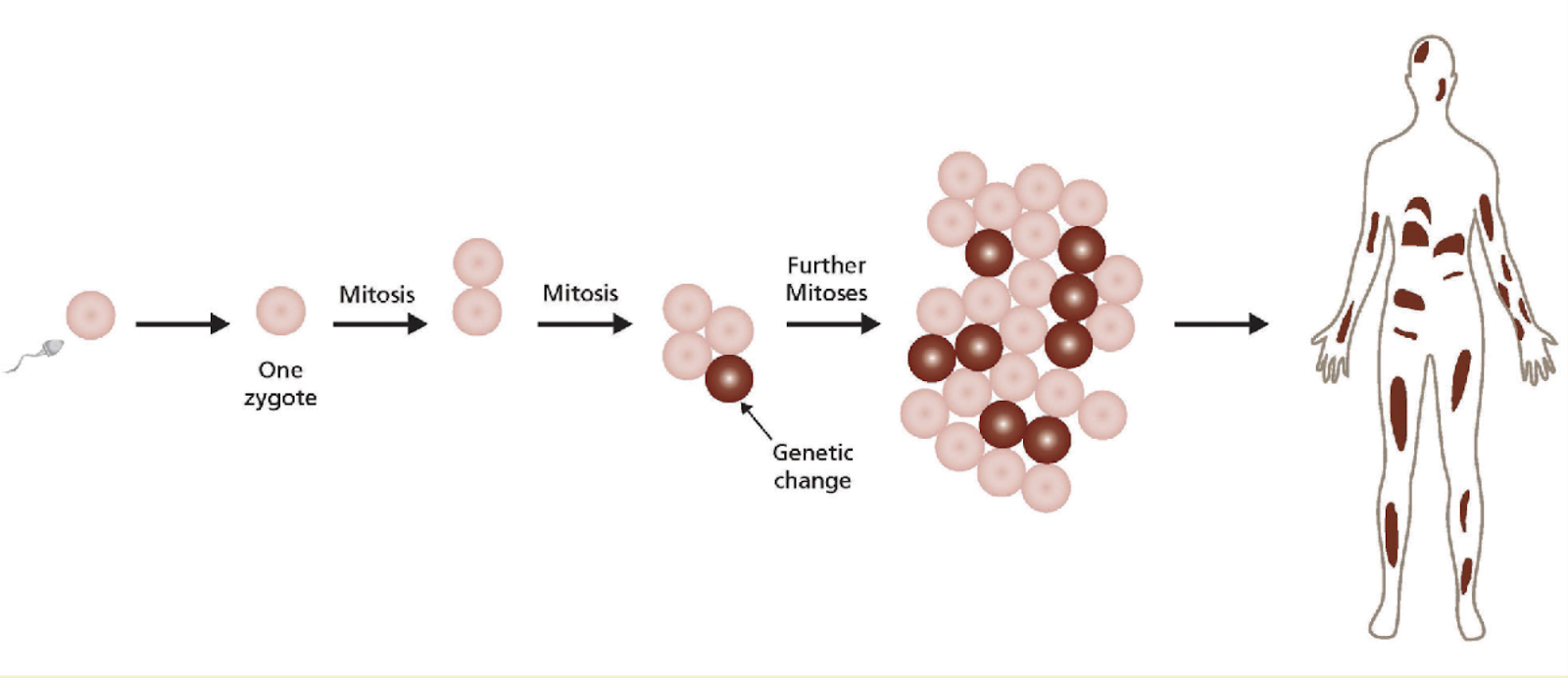
Mosaicism is the condition where a person has two or more genetically different sets of cells in his or her body. Sometimes, these different sets of cells may be aneuploid(1) [1]. Mosaicism derives from genetic changes in the DNA of our cells; for instance, the result of spontaneous DNA mutations or reversions of an existing DNA mutation, epigenetic changes in chromosomal DNA, as well as other abnormalities. These mutated cells then pass on to their daughter cells, creating a lineage (See Fig.1). There are also cases where these changes occur in mitochondrial DNA [2]. Moreover, mutation susceptibility increases when there is a higher genome instability due to direct and inverted repeats, the presence of DNA sequence characteristics (e.g. CpG dinucleotides), and the exposure to exogenous mutagens such as tobacco smoke - a chemical which particularly leads to a high prevalence of somatic mosaicism [3].
There are many different types of Mosaicism, the two main ones being Germline Mosaicism and Somatic Mosaicism. Germline Mosaicism, also known as gonadal mosaicism, is the case of mosaicism within gamete cells (as the name suggests). It can be caused by a mutation that takes place after conception or by epigenetic changes. As this happens early in development, it can be passed on to its daughter cells which can later be specialised into gametes. Therefore, a couple who may have mutations within their gamete cells may have children who suffer from certain genetic disorders such as Duchenne muscular dystrophy and osteogenesis imperfecta [4]. Mutations are recurrent in human cleavage-stage embryos as single-cell genomic analysis revealed that the chromosome instability is common in these young clusters of fertilised cells, and the abnormal chromosome complements can be found in approximately 70% of 14 normally developing human embryos that were examined [3].
However, in this article, we will mainly focus on Somatic Mosaicism; the case of mosaicism in our body cells (excluding the germline cells) that originate post-zygotically and apparently arising de novo during embryogenesis.
Some disorders associated with Mosaicism vary in severity depending on the timing of the first genetic change or mutation. The earlier the change, the larger the proportion of body cells affected. It also increases the possibility that it will become present in both somatic and germline cells in the future (which could potentially be passed on to future offspring, as mentioned in the previous paragraph). The genetic changes occurring later in development are more likely to stay as somatic mosaicism cases; those that are severe have a short window during development in which they must occur to be observed in adults [7]. However, there are instances when they don’t have to develop in this short window. At any stage in life, mutations in genes encoding tumour suppressors can be inactivated, or mutations in oncogenes may be activated, which can result in phenotypic effects in adults. An example of somatic mosaicism would be Cancer, which I will mention in the next paragraph. With this exception, Individuals with Somatic Mosaicism exhibit a milder phenotype due to the fact that only a proportion of cells have the mutation or because the mutation is being limited to a finite segment of the body [5].
Many well-known disorders have demonstrated somatic mosaicism, including Cancer, Down syndrome and neurofibromatosis (NF). Cancer is an example of somatic mosaicism because a cancer cell has a mutation in the genes responsible for replication and death of the cell, and this mutated cell has a different genetic makeup to the rest of the body cells. Studies have shown that approximately 2-4% individuals with Down syndrome are mosaic for cells that have three copies of chromosome 21 (i.e. some cells don’t have that third copy, whereas others do) [4]. Neurofibromatosis, a genetic disorder that results in tumours to form on nerve tissue (brain, spinal cord and nerves) can also be found in the mosaic form, which is known as segmental NF [5] [6]. Moreover, there are conditions of varying skin pigmentation and patterns that are the result of Somatic Mosaicism, and many form along the Blaschko Lines(2).
In this article, I will look into other cases of Somatic Mosaicism that are not as prevalent: Sturge-Weber Syndrome and Proteus Syndrome.
Sturge-Weber Syndrome
Sturge-Weber Syndrome (SWC), also known as encephalotrigeminal angiomatosis, is a sporadic congenital neurocutaneous disorder characterised by a port-wine stain that affects the skin in the distribution of the ophthalmic branch of the trigeminal nerve. The syndrome results in abnormal capillary venous vessels in the leptomeninges of the brain and choroid, and may result in conditions such as glaucoma, seizures, stroke and intellectual disability [8]. Studies have shown that in a group of people with port-wine stains (note that this includes patients that are not diagnosed SWC as port-wine stains are present in other disorders, but they all share the somatic mosaic mutation in the GNAQ gene), approximately 10% develop glaucoma and this is due to the high pressure on the optic nerve due to the malfunctioned capillaries [10].
SWC is caused by a somatic mutation in the GNAQ gene that occurs after the fertilisation of the embryo (p.Arg183Gln mutations). It is associated with somatic mosaicism because affected individuals have some cells with a normal copy of the gene and the rest with the abnormal gene (described as a ‘mosaic pattern’). This may be referred to as having two distinct cell lines in the body. The severity of SWC is therefore highly dependent on the ratio of healthy cells to abnormal cells in the body as well as the types of cells being affected [9].
The GNAQ gene codes a protein called Gαq that controls specific cell functions, which includes the regulation of blood vessels. Although more research is needed to fully understand the disruption of Gαq function in individuals, recent studies have shown that somatic mutation in GNAQ is enriched in endothelial cells which line the inside of blood vessel walls. This supports the association with the main symptoms of SWC (abnormal development of certain blood vessels), and these in turn can also cause other secondary effects to the affected tissue e.g. hypoxia(3), ischaemia(4), venous occlusion(5), thrombosis(6) and infarction(7). Calcification of affected areas of the brain may also occur [9].
Mutation in the GNAQ gene doesn’t only lead to SWC, but can also cause uveal melanoma (melanoma that affects the eye) which affects specific cells called melanocytes [9].
Treatment for SWC is specific for each symptom that the patient experiences. No treatment is required for Port-wine stains however many people choose to have them faded for cosmetic reasons, and this uses laser treatments that use a pulsed dye laser [10]. The laser treatment works by thinning the abnormal blood vessels so that the port-wine stain may fade or be removed. Seizures are treated with anticonvulsant medications however the effectiveness of this is variable as some patients do not respond to these medications. Hemispherectomy(8) is also an option for individuals affected by SWC because in some cases half of the brain is repeatedly damaged by chronic seizure activity. Other specific treatments include Focal cortical resection, Vagus nerve stimulation and preventive (prophylactic) treatment [9].
Proteus Syndrome
Proteus Syndrome is an extremely rare disorder that is characterised by the overgrowth of various tissues of the body. It is caused by a somatic activating mutation (mosaic variant) in the growth regulatory gene called AKT1 [11] [12] (medically known as the AKT1 c.49G→A variant) [11], which in turn results in random patches of disproportionate and asymmetric overgrowth around the body. Symptoms include progressive skeletal malformations, benign and malignant tumours, vascular malformations, bullous pulmonary disease and certain skin lesions. The disorder may become life-threatening as the abnormal blood vessels may increase the risk of deep vein thrombosis and pulmonary embolism [12].
Akin to Sturge-Weber Syndrome, the mosaic variant in AKT1 arises after the fertilisation of the embryo and people with Proteus Syndrome have some cells with a normal copy of this regulatory gene and some with the abnormal genes. The severity of Proteus Syndrome depends on the ratio of healthy cells to abnormal cells. In the scenario where all the cells have the abnormal gene, the condition is incompatible with life [12].
Only 200 patients have been reported to have Proteus Syndrome in medical literature. One of the first ones reported was a man called Joseph Merrick, an Englishman who lived in the 19th century, and many doctors in the past mistook his diagnosis for neurofibromatosis [11] [12]. This shows how a confirmed diagnosis of Proteus Syndrome is hard to reach as DNA diagnostic testing must be performed on the biopsies of affected tissues, and other techniques e.g. X-rays, CT Scans for skull lesions(9) and pulmonary cysts in the lungs and MRI of the brain, abdomen, pelvis and limbs are needed. Ultrasound is also used to evaluate the possibility of deep vein thrombosis in Proteus Syndrome patients [12].
In terms of treatment for Proteus Syndrome, multiple orthopaedic procedures (e.g. Epiphysiodesis) are needed to try and control the rapid overgrowth of cells and surgery may be required when overgrowth interferes with joint function or causes scoliosis or angular deformities. Individuals with Proteus Syndrome also need to have a checkup by a hematologist every few weeks to reduce risk of blood clots e.g. deep vein thrombosis [13].
Conclusion
Effectively, we are all mosaics as the 37 trillion cells in our body undergo mutations throughout our lifetime. Studies at Pennsylvania State University have shown that the average mammalian genome mutation rate is approximately 2.2 × 10-9 per base per year [14]. I feel as though a lot of the time, we tend to focus on the genetic variability between us and everyone else, but it’s important to realise that there is genetic variety within us. I hope that through reading this article, you have opened your eyes to the boundless possibilities our genetic self can have, as well as the importance in learning more about these rare ‘mosaic’ diseases in the hopes of potentially finding a way to modify any of these life-threatening conditions; however, at the same time, this could bring about ethical repercussions in the topic of human cell modification - posing a question on ways we can work around this. With ongoing developments, the answer is coming soon.
Glossary
Aneuploid: The presence of an abnormal number of chromosomes in a cell.
Blaschko Lines: Lines that represent pathways of epidermal cell migration and proliferation during the development of the fetus.
Hypoxia: a condition in which the body or a region of the body is deprived of adequate oxygen supply at the tissue level.
Ischaemia: inadequate blood supply to an organ or part of the body, especially the heart muscles.
Venous occlusion: a blockage of the small veins that carry blood away from an organ.
Thrombosis: formation of a blood clot, known as a thrombus, within a blood vessel.
Infarction: obstruction of the blood supply to an organ or region of tissue, typically by a thrombus or embolus, causing local death of the tissue.
Hemispherectomy: The removal or disabling of half the brain.
Lesions: regions in an organ or tissue which has suffered damage through injury or disease e.g. wound, ulcer, abscess, or tumour.
Further Reading
Somatic Mosaicism and Disease:
https://www.sciencedirect.com/science/article/pii/S0960982214005466
Genetic Mosaicism and Cancer: Cause and Effect:
Bibliography
[1] “Medical Genetics: Mosaicism.” Content - Health Encyclopedia - University of Rochester Medical Center, www.urmc.rochester.edu/encyclopedia/content.aspx?ContentTypeID=90.
[2] Chial, Heidi. “Somatic Mosaicism and Chromosomal Disorders.” Nature News, Nature Publishing Group, 2008, www.nature.com/scitable/topicpage/somatic-mosaicism-and-chromosomal-disorders-867/.
[3] Lupski, James R. “Genome Mosaicism—One Human, Multiple Genomes.” Science, vol. 341, no. 6144, 2013, pp. 358–359., www.jstor.org/stable/23491102. Accessed 11 Sept. 2020.
[4] “Germline Mosaicism.” Content, hihg.med.miami.edu/code/http/modules/education/Design/CoursePageContent.asp?ID=14.
[5] “Somatic Mosaicism.” Content, hihg.med.miami.edu/code/http/modules/education/Design/CoursePageContent.asp?ID=15.
[6] “Neurofibromatosis.” Mayo Clinic, Mayo Foundation for Medical Education and Research, 8 Apr. 2020, www.mayoclinic.org/diseases-conditions/neurofibromatosis/symptoms-causes/syc-20350490.
[7] Freed, Donald, et al. “Somatic Mosaicism in the Human Genome.” Genes, vol. 5, no. 4, 2014, pp. 1064–1094., doi:10.3390/genes5041064.
[8] Shirley, Matthew D., et al. “Sturge–Weber Syndrome and Port-Wine Stains Caused by Somatic Mutation in GNAQ.” New England Journal of Medicine, vol. 368, no. 21, 2013, pp. 1971–1979., doi:10.1056/nejmoa1213507.
[9] “Sturge Weber Syndrome.” NORD (National Organization for Rare Disorders), 23 June 2017, rarediseases.org/rare-diseases/sturge-weber-syndrome/.
[10] Iftikhar, Noreen. “Port-Wine Stains: Symptoms, Causes, Best Treatment Options.” Healthline, Healthline Media, 17 Mar. 2020, www.healthline.com/health/port-wine-stains.
[11] Lindhurst, Marjorie J., et al. “A Mosaic Activating Mutation in AKT1 Associated with the Proteus Syndrome: NEJM.” New England Journal of Medicine, 18 Aug. 2011, www.nejm.org/doi/full/10.1056/NEJMoa1104017.
[12] “Proteus Syndrome.” NORD (National Organization for Rare Disorders), 22 May 2018, rarediseases.org/rare-diseases/proteus-syndrome/.
[13] “Proteus Syndrome: Frequently Asked Questions.” Genome.gov, www.genome.gov/27544873/faq-about-proteus-syndrome.
[14] Kumar, S., and S. Subramanian. “Mutation Rates in Mammalian Genomes.” Proceedings of the National Academy of Sciences, vol. 99, no. 2, 2002, pp. 803–808., doi:10.1073/pnas.022629899.


Comments
Post a Comment